







患者の臨床的特徴
2015年から2017年までに合計177人の患者(男性81人、中央値 [range] 年齢、4 [0–30] 年)169家族のうちTOKAI-IRUDプログラムに依頼されました。 この研究に登録されたすべての患者は、新しい患者、すなわち以前に包括的なゲノム変異について分析されていない患者でした。 しかし、いくつかの患者がいくつかのフォローアップ調査に含まれています。19、20、21,22。
TOKAI-IRUDプログラムは、すべての患者を収容する可能性が開かれています。 志願者の臨床症状は、全発達遅延(HP:0001263; n = 95、54%)、発作(HP:0001250; n = 40、23%)、知的障害(HP:0001249; n = 29、16%)でした。 。 )、筋肉緊張の低下(HP:0001252; n = 24、14%)、奇形の顔の特徴(HP:0001999; n = 17、9.6%)、低身長(HP:0004322; n = 14、7.9%)、小頭症:0000252、n = 11、6.2%)およびその他(n = 38、21%)(表1、補足表S2および補足表S3)。
同定された亜種
ACMG指令によれば、36人(20%)の患者で病原性SNVが同定された。 さらに、30人(17%)の患者が臨床的妥当性評価と臨床情報と適用可能な疾患表現型の一貫性とに基づいて「病原性の可能性がある」と分類されたSNVを保有しました。 病原性または病原性SNVを有する66人の患者のうち、47人は常染色体優性遺伝疾患、7人は常染色体劣性遺伝疾患、8人はX関連優性遺伝疾患、4人はX関連劣性遺伝疾患を有していた(図1)。
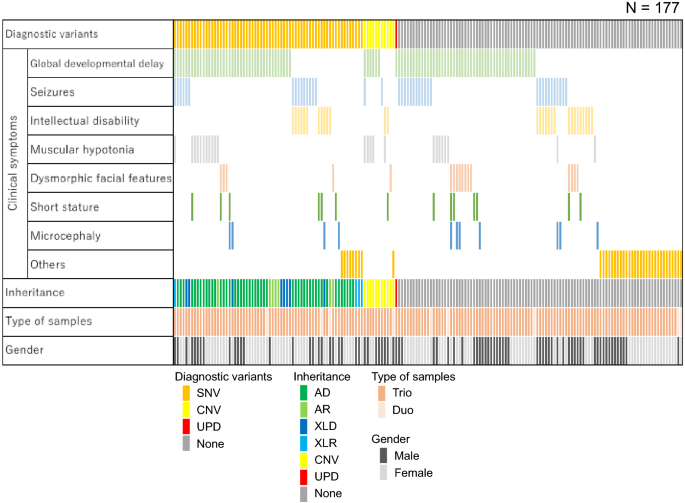
検出された変異体の患者特性および情報。 各列は1人の患者を表す。 SNV 単一ヌクレオチド変異体、 CNV コピー番号バリアント、 UPD 一方的な二染色体、 起源の後 常染色体優性、 AR 常染色体劣性、 XLD X-接続優性、 XLR X連想劣性。
コピー数分析は、11人の患者(6.2%)で診断重複/削除を確認し、ここでは10q26.3削除(TOKAI-IRUD-1135およびTOKAI-IRUD-1273)、22q11.2重複(TOKAI-IRUD-1236) 、5q14.3削除(TOKAI-IRUD-1252)、47、XXY(TOKAI-IRUD-1297)、1p36削除(TOKAI-IRUD-1301)、7q11.23重複(TOKAI-IRUD-1321)、19p13.1 (TOKAI-IRUD-1335)、16p13.3重複(TOKAI-IRUD-1337)、17p11.2重複(TOKAI-IRUD-1343)、および4p16.3重複(TOKAI-IRUD-1475)。
ROH分析は、105のケースで10Mbを超えるホモ接合領域を同定した。 これには、Angelman症候群(TOKAI-IRUD-1290、OMIM#105830)と診断された1人の患者(0.6%)で診断upd(15)patが含まれていました。 さらに、全染色体のUPDが2人(1.1%)の患者で確認されました。 [upd(2)pat; TOKAI-IRUD-1249 and upd(3)pat; TOKAI-IRUD-1180] 診断SNVまたはCNVはありません。 これにより、177人の患者のうち78人(44%)について遺伝子診断を受け、そのうち10人(13%)が本事業が始まった2015年以降に認知された疾患と診断された。 かなりの数の患者がより軽い表現型を示した(26 [33%])、より深刻な表現型(9 [12%])、または非定型複合表現型(17 [22%])各疾患の既存の臨床的表現と比較する。
広範なUPD領域を持つ患者の症例を発表
upd(15)patを持つTOKAI-IRUD-1290:サンプルを提出したときに2歳の少年である患者は、血縁ではなく健康な親の3人の子供の3人目でした(図2b)。 胎児期から疑われるイランイ形成症は、出生後磁気共鳴映像撮影(MRI)で確認した(図2a)。 経口摂取が難しく、管栄養を行い、繰り返しの吸引性肺炎で気管切開術を施行した。 彼はまた、先天性受信腎症、先天性甲状腺機能低下症、胃食道逆流疾患、発達遅延、てんかん、難聴、喉頭臓器軟化症を患っていた。 ROH分析は、染色体15の長腕の全長にわたって父系UPD領域を同定した。 [upd(15)pat]地域を含む UBE3A Angelman症候群(OMIM#105830)の診断につながった遺伝子(図2b)。 さらに、11個のホモ接合性レア変異体が父系由来UPD領域で同定されており、これには以下が含まれます。 デュオックス2 (c.G1560C、p.E520D)バリアント。 デュオックス2 は先天性甲状腺機能低下症の原因遺伝子として知られていますが、この特定の変異は以前に報告されていません。
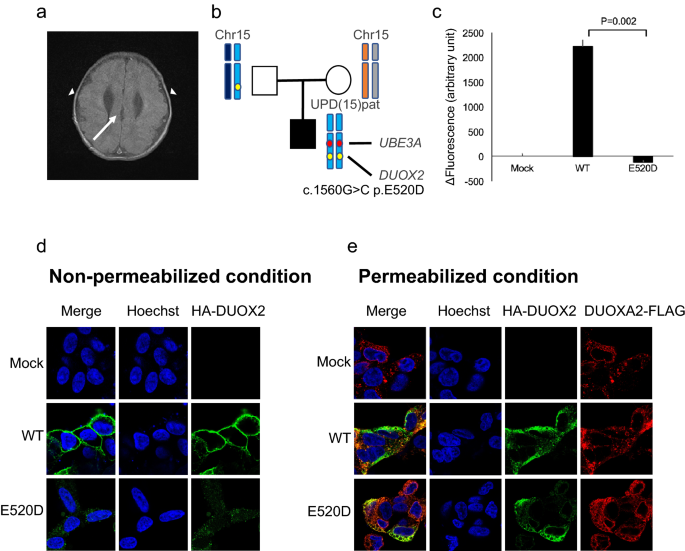
TOKAI-IRUD-1290の臨床的特徴とUPD分析の結果 (ㅏ) 側頭葉の皮質異形性症(矢印)と脳量形成異常(矢印)を示す2歳の脳MRI。 (非) UPD分析の結果。 15番染色体長腕の全長にわたる父系遺伝UPD領域 [upd(15)pat] UBE3A遺伝子領域を含むことが確認された。 (氏) 時間2永久2– DUOX2タンパク質の生産能力は、共発現されたDUOXA2-FLAGの存在下でAmplex Red試薬によって測定された。 変異体の活性は、WT(100%)および模擬トランスフェクト対照(0%)の活性に基づいて標準化された。 データは、同様の結果を持つ3つの独立した実験(それぞれ3回実行)を表します。 Tバーは平均の標準誤差を表します。*血< 0 05対WT(Welch's ティー-テスト)。 (D) HAタグ付きDUOX2構築物(WTまたはE520D、緑色蛍光)を用いた細胞内位置解析。 (金利) 透過性条件下での蛍光免疫染色は、E520D-DUOX2の位置がDUOXA2と一致することを示した。
この場合、検出されたDUOX2 p.E520Dミスセンス置換の病原性を確認するために、HEK293細胞を用いて発現実験を行った。2永久2– 同時発現されたE520D突然変異体の生産能力 デュオキサ2-FLAGが評価されました。 我々は、E520D突然変異がHの完全な損失を示すことを示しています2永久2-生産活動(図2c)。 免疫蛍光を用いた細胞内局在化の可視化は、WTとE520D突然変異体(図2d、e)の間の膜発現レベルで有意な差を示した。
upd(3)pat を持つ TOKAI-IRUD-1180: サンプル提出当時 3 歳の少女であるこの患者は、健康な血縁ではなく両親の一人娘でした。 彼女は出生後1日目から発作を起こし、脳波検査で異常所見が発見され、症状性てんかんが疑われました。 しかし、フェノバルビタールの経口投与が始まった14日目から発作が止まった。 彼女は座ることができず、サンプルを提出したときに言語の理解が悪かった。 ROH分析では、染色体3の全長UPDが明らかになりました。 [upd(3)pat]3番染色体で40個の同型接合レアミスセンス変異が確認されたが、WES分析では遺伝的診断には到達できなかった。
upd(2)pat を持つ TOKAI-IRUD-1249: サンプルの提出の時点で 4 ヶ月の少女である患者は、健康な血縁ではなく親の一人娘でした。 出生前のMRIで水頭症が確認された。 彼女は妊娠34週に予定された帝王切開手術で生まれ、難聴、両側湾曲族、両側股関節脱臼、多発性関節構築、先天性水頭症、心室中隔欠損、発達遅延、短く緩やかな大腿骨、鐘状の胸郭を患っていました。 および外の開口部のない質。 ROH分析により、染色体2の全長UPDが明らかになった。 [upd(2)pat]。 彼女は生後10ヶ月で吸引性肺炎で死亡し、2番染色体で希少同型接合ミスセンス変異34個とナンセンス変異1個が確認されたが、WES分析は遺伝的診断につながなかった。
付随的発見
付随的発見を報告するためのACMG勧告に含まれる遺伝子の1つの病原性変異体が、1人の患者(TOKAI-IRUD-1150)、すなわちc.C6952Tにおいて検出された。 BRCA2。 さらに、3人の家族で矛盾する親と子供の関係が確認されました。






+ There are no comments
Add yours