






患者サンプル
Kyoto Prefectural University of MedicineでSTZで治療された8人のNET患者を後向きに登録しました。 すべての臨床特性は、年齢、性別、合併症および実験室結果を含む医療記録から収集され、補足表1にリストされています. ヒト腎臓サンプルは、腎生検を介して2人の患者から得られた。 対照として、管状組織ではなく糸球体に主に関与する薄い基底膜疾患を有する患者の腎臓組織を分析した。 患者情報は分析前に匿名化され、非識別された。 本研究の完全なプロトコルは、ヘルシンキ宣言に従って設計されています。 遡及設計と患者のリスクが低いため、倫理委員会は次のオプトアウト方法論の使用を承認しました。 口頭辞書同意の要件は免除され、一般にアクセス可能な情報と簡単なオプトアウトモードによって事前同意が得られました。 本研究は京都府立医科大学病院医療倫理委員会の承認を受けました(承認番号:ERB-C-2169、ERB-C-2210)。
動物実験
雄C57BL / 6野生型マウスはShimizu、Inc.(Kyoto、Japan)から購入した。 STZ損傷モデルは、0.05Mクエン酸ナトリウム緩衝液(pH 4.5)にSTZ(Cayman Chemical、MI、USA)を100、150、または200mg / kg(体重)の濃度で雄マウスに腹腔内注射することによって誘導した。 8-10週。 STZ注射の1日後または7日後にマウスを安楽死させた。 一匹の子コントロールマウスを群間比較に使用した。 全てのインスリン処理群は、インスリングラジン(Wako、Osaka、Japan)を皮下注射した後、空腹時血糖を同じレベル(±insulin glargine 2-6 Units / kg)に維持する量に調整した。
インビボでp53を阻害するために、前述のようにSTZ注射の直前にピピトリン-α(ab120478、Abcam plc.、Cambridge、UK)2.2mg/kgを腹腔内注射した。41。 インビボでSGLT2を阻害するために、1.0 mg/kgのダパクリフロジン(Selleck Biotech Co., Houston USA)を溶解し、75%生理食塩水(0.9% w/v NaCl)で希釈し、STZ処理の1時間前に胃管栄養法で投与する。しました。 体重と血糖値は8時間絶食後17:00に測定した。 血糖値は、血糖計(Glutest Every、Sanwa Kagaku Kenkyusho Co.、Ltd.、Aichi、Japan)を用いて測定した。 マウスをイソフルランで麻酔し、指定された時点で安楽死させた後、下大静脈から血液サンプルを採取した。 さらなる分析のために、腎臓および膵臓をサンプルに切断した。
すべての実験は、京都医学大学の実験動物委員会によって承認され、日本科学委員会およびARRIVE指令による適切な動物実験の実施に関する機関の指針および指針に従って行われた。
細胞培養と細胞生存率の分析
正常なラット腎臓上皮細胞(NRK 52E細胞)はJCRB細胞銀行から購入した。 細胞を、1%ペニシリンおよびストレプトマイシン(Invitrogen、Carlsbad、CA)および5%FBS(Invitrogen)を含むDMEM(Wako、Osaka、Japan)で37℃、加湿した5%CO 2で培養した。2 そして95%大気雰囲気。 細胞を計数する前に、24時間ジメチルスルホキシドに溶解した1、5、10または30mMのSTZで細胞を処理した。 細胞数は、死細胞染色のプロピジウムヨージド染色法を用いたADAM-MC自動細胞計数機(Digital Bio, Japan)を用いて計数した。
血清血液尿素窒素および血漿インスリン測定
血清血液尿素窒素(BUN)は、適切な酵素法(A667-00、Serotec、北海道、日本)を使用して測定されました。 収集された血漿中のインスリン濃度は、製造元の指示に従って、ウルトラセンシティブマウスインスリンELISAキット(Morinaga Institute of Biological Science、Inc.、金沢、日本)を用いて測定した。
組織の準備と組織学
マウスを麻酔および犠牲にし、示された時点で膵臓および腎臓を摘出した。 パラフィン切片を準備するために、膵臓と腎臓を4%パラホルムアルデヒドで固定し、Applied Medical Research Laboratory(Osaka, Japan)でパラフィンに包埋した。 パラフィン内蔵マウスおよびヒト組織を4μmの厚さに切断した。 PAS(Periodic Acid-Schiff)染色は標準手順に従って行った。
免疫組織化学
脱パラフィン処理後、ラットとヒトのパラフィン切片をクエン酸塩緩衝溶液(pH6.0)に入れ、5分間煮沸して抗原を回収した。 内因性ペルオキシダーゼは、メタノール中の3.0%過酸化水素で20分間クエンチされた。 試料を室温で30分間PBS中3%BSAでブロックし、一次抗体と共にインキュベートした(補足表2)。 切片をヤギ抗ウサギHRPコンジュゲート二次抗体(ab236469、Abcam)で標識した。 ジアミノベンジジン発色基質(K3468、Agilent Technologies、Inc.、Santa Clara、CA)を発色反応に使用した後、ヘマトキシリンで対比染色した。 すべてのセクションは、Eclipse E600顕微鏡(Nikon、Tokyo、Japan)およびBZ-X700 / BZ-X710顕微鏡(Keyence Corporation、Osaka、Japan)を使用して観察されました。 γH2AX陽性細胞は、高倍率(n = 3)で、各腎臓の10の連続した重複しない皮質および水質場の3つで定量化された。 これら3つのフィールドは、ブラインド方式でランダムに選択された。
免疫蛍光染色
免疫蛍光染色のために凍結切片を再水和し、PBS中0.5%Triton X-100で5分間透過させた。 試料をPBS中3%BSAでブロックし、4℃で一晩補充表2に示した一次抗体と順次インキュベートし、次いでアレクサ・フルオル594結合二次抗体(補足表2)と1時間インキュベートした。 DAPIまたはDRAQ5(DR50050;BioStatus、英国レスターシャー、1:2000)を用いて核対照染色を行った後、Prolong-Gold(Thermo Fisher Scientific)に取り付けた。 画像は共焦点顕微鏡(FV1000、Olympus、東京、日本)で得た。
RNA抽出とリアルタイム定量PCR
総RNAは、TRIzol(Life Technologies、Carlsbad、CA)およびDirect-zol RNA MiniPrep(Zymo Research、Irvine、CA)を用いて腎臓またはNRK-52E細胞の皮質から抽出した。 全RNAの200ナノグラムを逆転写し、gDNA Eraser(Takara Bio)を含むPrimeScript RT試薬キットを用いてcDNAを合成した。 PCR産物のリアルタイム検出は、KAPA SYBR FAST qPCR Master Mix(2x)Universal(Kapa Biosystems、Wilmington、MA)およびThermal Cycler Dice Real Time System(Takara Bio Inc.)を用いて行った。 内部対照としてβ-アクチンを用いて遺伝子発現を定量した。 プライマーを補足表3に示す。
RNAシークエンシング
STZ群のマウス近位尿細管上皮細胞株であるNRK52E細胞を、RNA抽出前に24時間、10mMのSTZで処理した。 RNAサンプルは、ライブラリーの構築と配列決定のために、大阪大学微生物病研究所のゲノム情報研究センターのNGSコア施設に提供されました。 製造元の指示に従って、TruSeq鎖mRNAサンプル調製キット(Illumina、San Diego、CA)を使用してライブラリー調製を行った。 シーケンシングは、Illumina HiSeq 2500プラットフォームで100bpシングルエンドモードで行った。 シーケンスされた読み取りはSTAR v 2.7.10a(https://github.com/alexdobin/STAR/)と一意にマッピングされた読み取りは、SubreadパッケージのfeatureCounts関数によって計算されました(https://subread.sourceforge.net)。
生物情報学的分析
データ分析はRソフトウェアバージョン4.1.2(https://www.R-project.org/)。 edgeR パッケージを微分表現分析に使用した。42。 FDR < 0.05 및 절대 log2 배수 변화 > 1人の遺伝子は、有意に差別的に発現される遺伝子と考えられています。 fgseaパッケージを遺伝子セット濃縮分析に使用した。 KEGG(Kyoto Encyclopedia of Genes and Genomes)経路解析(https://www.genome.jp/kegg/kegg1.html)はiDEPバージョン0.93(http://bioinformatics.sdstate.edu/idep93/)経路分析のための一般的に適用可能な遺伝子集合強化(GAGE)法43。
ウェスタンブロット分析
溶解緩衝液17(R&D Systems、Inc.、Minneapolis、MN、USA)を使用して全細胞または組織抽出物を得た。 タンパク質を95℃で5分間加熱して変性し、SDS-PAGEで分離した。 次にタンパク質をポリビニルリデンジフルオリド膜(Immobilon-P IPVH00010:Millipore、MA、USA)に移した。 室温で1時間、TBS/0.1%Tween20の5%無脂肪ミルクまたは3%BSAでブロックした後、膜を4℃で一晩一次抗体(補足表2)と共にインキュベートした。 TBS/0.1%Tween20で洗浄した後、二次ペルオキシダーゼ結合二次抗体を添加した(7074S; Cell Signaling Technology, Boston, MA;室温で1:3000)。 ECL選択ウェスタンブロット検出試薬(RPN2235:GE Healthcare UK Ltd、Amersham Place、England)またはClarity Max western ECL基質(1,705,062:Bio-Rad Laboratories、Inc.、Hercules、CA、USA)を使用して化学発光を検出した。 。 ImageJソフトウェア(National Institutes of Health、Bethesda、MD)を使用して信号強度を評価した。
彗星分析
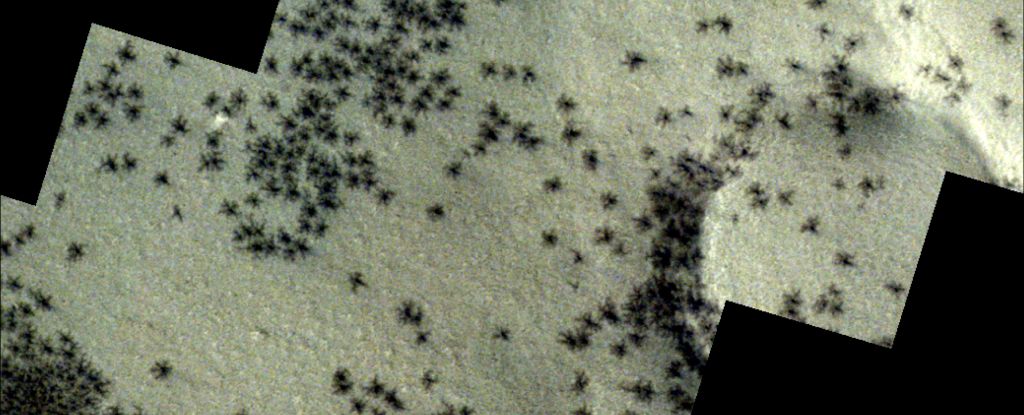
彗星分析は、前述のようにComet Assay Kit(Abcam ab238544)を用いて行った。41,44。 簡単に言えば、マウスの腎臓を取り除き、20mM EDTAを含む少量の氷のように冷たいPBSで細かく切った。 上清を35μmセルストレーナーに通した。 遠心分離後ペレットを1×105 氷のように冷たいPBSからcells/ml。 サンプルをComet Agaroseと1/10比(v / v)で混合し、Comet Agaroseベース層で覆われたガラススライドに移した。 予め冷却した溶解緩衝液と共にインキュベートした後、スライドを電気泳動した。 Alkaline comet assayはAlkaline Electrophoresis Solution、neutral comet assayはTBE Electrophoresis Solutionで電気泳動を行った。 電気泳動後、スライドをVista Green DNA染料と共にインキュベートした。 FITCフィルターを用いて、エピフルオロセンスマイクロスコープ(IX71;オリンパス、東京、日本)で画像を得た。 10枚の写真(写真あたり5〜15細胞)をランダムに撮影し、Comet Score分析ソフトウェア(TriTek Corp.)を使用して、グループあたり100細胞の尾モーメント(尾長×尾%DNA / 100)を計算した。
統計分析
結果は平均±標準誤差(SE)で表されます。 統計分析は対になっていない人によって行われた。 ティー– 2つの変数の比較と分散分析のためのテストと複数の変数の比較のためのDunnettの事後テスト。 血 0.05未満の値を有意とみなした。




+ There are no comments
Add yours